
pplacerの使い方
pplacerとは
pplacerはメタゲノムデータ中にどのような種が多いかを可視化・比較するツールです.
メタゲノム中に含まれる遺伝子配列ひとつひとつについて,予め作成したリファレンス系統樹(16Sなどのuniversal geneの遺伝子系統樹)上のどこに配置(placement)するべきかを計算し,メタゲノムデータに含まれる種の構成を系統樹上の枝の太さに変換します.
全体の流れ
矢印の先に使用するソフトウェアを書いてあります.
- 必要なツールのインストール
- あるオーソログについての配列を集める(リファレンス配列)
- リファレンス配列からマルチプルアラインメントを計算する(リファレンスアラインメント) → MAFFTなど
- マルチプルアラインメントから系統樹を推定する(リファレンス系統樹) → FastTreeまたはRAxML/phyml
- 上記で作られたファイルから,リファレンスパッケージを作成する → taxtastic
- あるオーソログに類似した配列をメタゲノムデータから集める → BLAST, HMMER, Infernalなど
- リファレンスアラインメントに対してメタゲノムのリードをアラインメントする → MAFFTなど
- メタゲノムのリードをリファレンス系統樹へ配置(placement)する → pplacer
- 可視化,比較などを行う → guppy, Archaeopteryx
mothur/QIIMEユーザ向け情報
pplacerのアップデート報告によると,mothur/QIIMEのBIOM fileとtreeをリファレンスパッケージと.jplaceファイルの代わりに使えるそうです.
フォーマットの説明
## リファレンスパッケージ(Reference package)の作成に必要なファイル
seqs.fa: リファレンス配列のマルチプルアラインメント.FASTAフォーマット.tree.nwk: リファレンスアラインメントから作成された系統樹.Newickフォーマット.tree_stat.txt: FastTreeのログファイル,または,RAxML/phymlのstatisticsファイル.
さらに,オプションで追加できるものがあります.詳しくはこちらを参照してください.
tax_ids.txt: リファレンスアラインメントに含まれる配列のtaxon id のリスト.一行ごとにIDが書かれているテキストファイル.seq_info.csv: アラインメントされた配列に関するCSVファイル. ヘッダーはseqname,accession,tax_id,species_name,is_typeとする.一行ごとに,マルチプルアラインメントに含まれる配列について記述する.最低限,seqname,tax_idがあればよい.
リファレンスパッケージ(Reference package)
- リファレンスアラインメント,リファレンス系統樹,分類学的情報に関するファイルを集めたディレクトリ(*.refpkg)
taxtasticを用いて作成する- リファレンスパッケージの例
プレイスファイル(Place file)
- リファレンス系統樹上へのリードの配置(placement)に関する情報
- JSONフォーマット(*.jplace)
必要なツールのインストール
## MAFFTのインストール
MAFFTはマルチプルアラインメントを高速に行うツールです.
こちらからダウンロードできます.
FastTreeのインストール
FastTreeはマルチプルアラインメントから系統樹を推定するツールです.
Downloading and Installing FastTreeを参考にしてインストールしてください.
# Macの場合
gcc -O3 -finline-functions -funroll-loops -Wall -o FastTree FastTree.c -lm
taxtasticのインストール
taxtasticはリファレンスパッケージの作成するツールです.リファレンスパッケージは,リファレンス系統樹,リファレンスアラインメント,プロファイル,およびその他のメタデータから構成されるディレクトリです.
taxtastic/README.rstを参考にしてインストールしてください.
# easy_installによるインストール
sudo easy_install taxtastic
pplacerのインストール
LinuxまたはMax OSXに対応しています.こちらのページからバイナリファイルをダウンロードしてください.
ダウンロードした圧縮ファイルを解凍すると,ディレクトリにguppy,pplacer,rpprという3つのプログラムがあると思います.システムがそれらのプログラムを見つけられるように,このディレクトリにパスを通してください.
Archaeopteryxのインストール
Archaeopteryxは系統樹の可視化ツールです.こちらのページから,forester.jarをダウンロードしてください.
また,_aptx_configuration_fileというconfiguration fileをこちらのページからダウンロードしてください.
リファレンスパッケージの作成まで
## あるオーソログについての配列を集める
16SrRNAの場合はInfernalなどの二次構造を考慮した配列検索ツールを,タンパク質コード遺伝子であればBLASTやHMMERなどを利用することになると思います.ここでは詳しくは触れません.
# ここではテストデータを作成します(50個のエントリを含むFASTAファイル)
$ head -n 100 GreenGenesCore-May09.ref.fna > seq.fa
リファレンス配列からマルチプルアラインメントを計算する
MAFFTでマルチプルアラインメントを計算します.
$ mafft --auto seq.fa > seq_align.fa
マルチプルアラインメントから系統樹を推定する
FastTreeで系統樹の推定します.この際,-logオプションでログファイルを出力します.
FastTree -gtr -nt -log fasttree.log < seq_align.fa > tree.nwk
リファレンスパッケージの作成
-lオプションで任意の遺伝子名を表す名前を付けることができます.-Pオプションで出力するリファレンスパッケージの名前を指定します.
# リファレンスパッケージの作成
taxit create -l 16s_rRNA -P my.refpkg --aln-fasta seq_align.fa --tree-stats fasttree.log --tree-file tree.nwk
また, 以下のように,分類情報を含むリファレンスを作成することもできます.
# NCBI taxonomyのデータをtaxonomy.dbとしてダウンロードする
taxit new_database -d taxonomy.db
# tax_ids.txtの作成
awk '/>/{sub(/>/,""); print }' seq.fa > tax_ids.txt
# tax_ids.txtに含まれる全ての種についてminimum sub taxonomyを格納した taxa.csv を作成する
taxit taxtable -d taxonomy.db -t tax_ids.txt -o taxa.csv
# リファレンスパッケージの作成
taxit create -l locus_name -P my.refpkg --taxonomy taxa.csv --aln-fasta seqs.fasta --seq-info seq_info.csv --tree-stats tree_stats.txt --tree-file tree.nwk
リファレンスパッケージの作成に必要なファイルのフォーマットについては,taxtasticのQuickstartを参照してください.
リファレンスパッケージを作成してから
## あるオーソログに類似した配列をメタゲノムデータから集める BLAST, HMMER, Infernalなどを使って,抽出してください
リファレンスアラインメントに対してメタゲノムのリードをアラインメントする
-addオプションで追加するメタゲノムのリードを指定します.
mafft --auto --add test.fa seq_align.fa > test_align.fa
また,リファレンス配列とメタゲノムデータ由来の配列を混ぜた(マージした)配列について,MAFFTを実行することも可能だと思います.
メタゲノムのリードをリファレンス系統樹へ配置(placement)する
# Place file の作成(*.jplaceがつくられる)
pplacer -c my.refpkg test_align.fa
可視化
guppyで可視化のために使うphyloXMLフォーマットのファイルを作成します.
# phyloXMLフォーマットのファイルの作成(リファレンスパッケージに分類情報が含まれる場合)
guppy fat -c vaginal_16s.refpkg p4z1r36.jplace
# phyloXMLフォーマットのファイルの作成(リファレンスパッケージに分類情報が含まれない場合)
guppy fat test_align.jplace
リファレンスパッケージに分類情報が含まれないときに,guppy fatの-cオプションでリファレンスパッケージを指定すると,以下のようなエラーが出ます.この場合は,-cオプションを使用しないとうまくいきます.しかし,可視化の際に分類情報は表示されません.
$ guppy fat -c my.refpkg test_align.jplace
Uncaught exception: Refpkg.Missing_element("taxonomy")
Fatal error: exception Refpkg.Missing_element("taxonomy")
phyloXMLファイルができたら,archaeopteryxを実行します.
java -jar forester.jar -c _aptx_configuration_file GreenGenesCore-May09.ref.align.xml
比較
guppy krは複数のplace file(.jplace)を比較し,サンプル間の距離行列を作成します.ここでの距離は,UniFracを一般化した,Kantorovich-Rubinstein (KR) metricです.KR metricは一つのplacement(リードの系統樹上での分布)から別のplacementに変換するのに要する移動の量と解釈できます.
# *.jplace はカレントディレクトリにある.jplaceファイルすべてを指定しています
guppy kr *.jplace
guppy kr_heatで2つのplace file(.jplace)を比較した図を作成することができます.枝上の移動量に比例して,枝を太くなります(赤は根に向かう方向,青は葉に向かう方向の移動を表す).つまり,2つのサンプル間で違いが大きい枝が太くなります.
guppy kr_heat -c foo.refpkg bar1.jplace bar2.jplace
java -jar forester.jar -c _aptx_configuration_file bar1.bar2.heat.xml &
Edge principal components analysis
guppy pcaで,placementを特徴量としたPCAによりサンプル間比較ができます.詳しくはこちらを読んでください.
guppy epca --prefix pca_out -c foo.refpkg *.jplace
java -jar forester.jar -c _aptx_configuration_file pca_out.xml &
pca_out.transにはPCAの軸へ射影されたサンプルの情報が含まれています.
Squash clustering
guppy squashで,placementに特化したサンプルの階層的クラスタリングを行えます.--out-dirオプションで指定したディレクトリ内にcluster.treというクラスタリングの樹形を格納したファイルを作成し,また,内部ノードごとの平均を格納したmass_treesというサブディレクトリを作成します.
mkdir squash_out
guppy squash -c foo.refpkg --out-dir squash_out *.jplace
java -jar forester.jar -c _aptx_configuration_file squash_out/cluster.tre &
クラスタリングの樹形図の6番という内部ノードについて可視化することができます.
java -jar forester.jar -c _aptx_configuration_file squash_out/mass_trees/6.phy.fat.xml &
メタゲノムのリードの分類
guppy classifyによって,メタゲノムのリードを分類できます.出力のカラムはread name, attempted rank for classification, actual rank for classification, taxonomic identifier, confidenceです.
guppy classify --mrca-class -c foo.refpkg bar.jplace > classify.txt
リードの分類をSQlite3で閲覧することもできるそうです.詳しくはpplacerのデモを読んでください.
デモ
## テストデータの作成
リファレンス配列はDDBJの16S rDNA配列から作成しました(ref.fa).また,リファレンス配列のtaxon idのリスト(taxid.txt),および,ref.faでの配列名とtaxon idを紐付けるcsvファイル(ref.csv)を作成しました.
# リファレンス配列の準備(100エントリのFASTA)
awk '{if(/>/){n++}; if(n>100){exit}; print}' 16S.seq > ref.fa
$ head taxid.txt
1334
81436
1318
1596
334344
76637
78164
198312
1520
2743
$ head ref_info.csv
"seqname","tax_id"
"AB002492","1334"
"AB003466","81436"
"AB006127","1318"
"AB008209","1596"
"AB010844","334344"
"AB012648","76637"
"AB015608","78164"
"AB017113","198312"
"AB020191","1520"
メタゲノムのリードはFastUniFracのチュートリアルにあったGreenGeneの16Sの配列から作成しました.
# メタゲノムのリードの準備(500エントリのFASTAファイル; 16S)
awk '{if(/>/){n++}; if(n>500){exit}; print}' GreenGenesCore-May09.ref.fna > seq1.fa
awk 'NR>1000{if(/>/){n++}; if(n>500){exit}; print}' GreenGenesCore-May09.ref.fna > seq2.fa
本番
# リファレンスアラインメントの計算
mafft --auto ref.fa > ref_align.fa
# リファレンス系統樹の推定
FastTree -gtr -nt -log fasttree.log < ref_align.fa > ref_tree.nwk
# リファレンスパッケージの作成
taxit new_database -d taxonomy.db
taxit taxtable -d taxonomy.db -t taxid.txt -o taxa.csv
taxit create -l 16s_rRNA -P my.refpkg --taxonomy taxa.csv --aln-fasta ref_align.fa --seq-info ref_info.csv --tree-stats fasttree.log --tree-file ref_tree.nwk
# リファレンスアラインメントへのメタゲノムのリードのアラインメント
mafft --auto --add seq1.fa ref_align.fa > seq_align1.fa
mafft --auto --add seq2.fa ref_align.fa > seq_align2.fa
# Place file の作成(*.jplaceがつくられる)
pplacer -c my.refpkg seq_align1.fa
pplacer -c my.refpkg seq_align2.fa
# 可視化
guppy fat -c my.refpkg seq_align1.jplace
guppy fat -c my.refpkg seq_align2.jplace
java -jar forester.jar -c _aptx_configuration_file seq_align1.xml &
このように可視化されます.
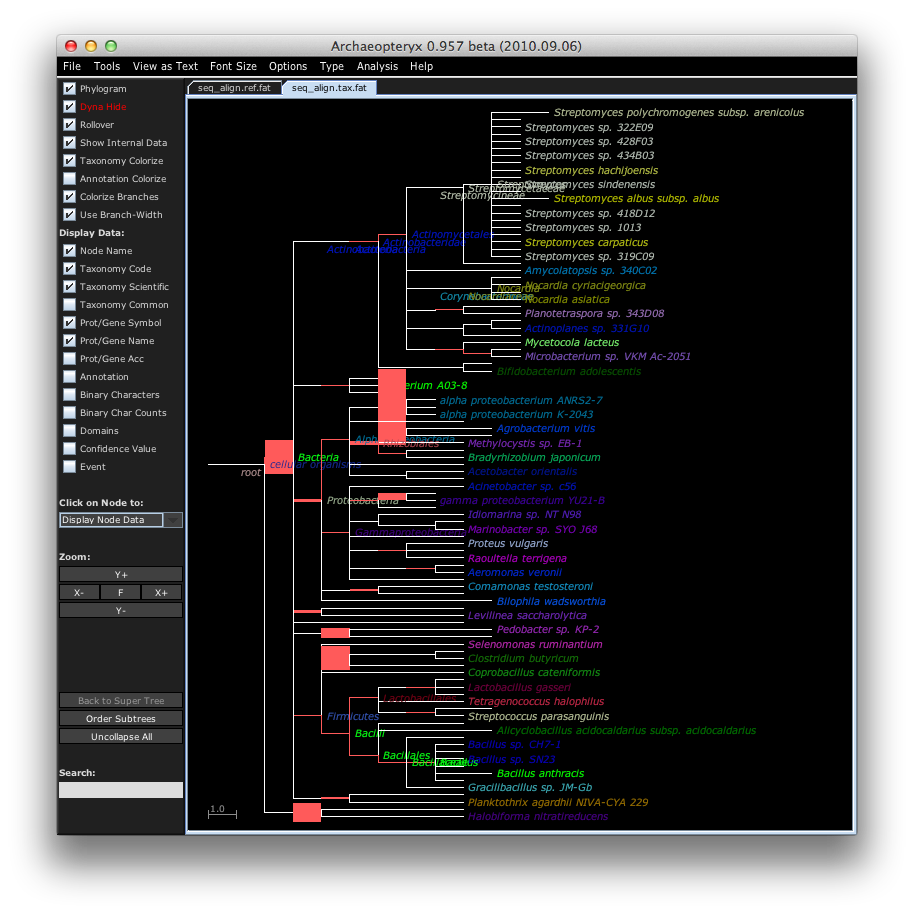
# サンプル間距離の計算
guppy kr seq_align1.jplace seq_align2.jplace
# 2サンプル間比較
guppy kr_heat -c my.refpkg seq_align1.jplace seq_align2.jplace
java -jar bin/forester.jar -c _aptx_configuration_file seq_align1.seq_align2.heat.xml &
このようになります.
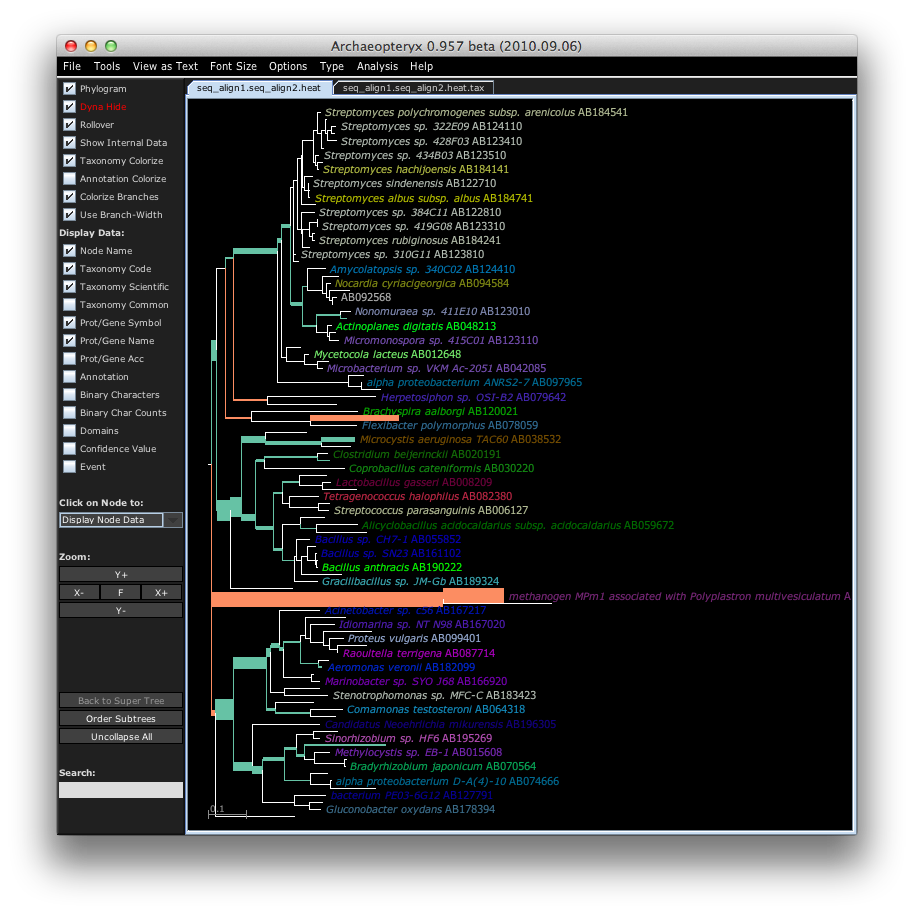
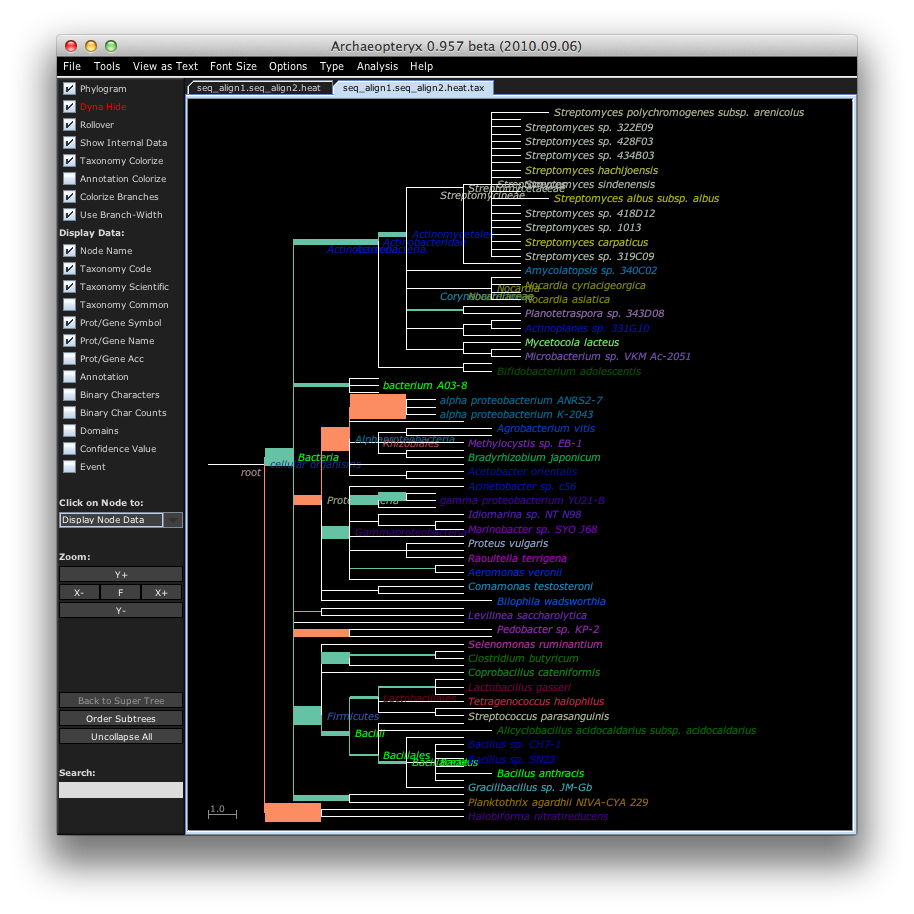
補足
## guppy (Grand Unified Phylogenetic Placement Yanalyzer)について
# サブコマンドの一覧を表示
guppy --cmd
# サブコマンドの実行(SUBCOMMANDの部分に使いたいサブコマンドを挿入する)
guppy SUBCOMMAND [options] [files]
# サブコマンドのヘルプ
guppy SUBCOMMAND --help