
MOCCS2とは?
MOCCS2 (MOCCS version 2.0) は、様々なChIP-Seqのデータから転写因子が認識する配列を正確に抽出できるバイオインフォマティクス手法 MOCCS を偽陽性が少なくなるように改良したソフトウェアです。
MOCCS2のソフトウェアはGitHubにて公開しています。 https://github.com/yuifu/moccs
私たちはMOCCS2を開発するとともに、時計タンパク質DBPおよびMOCCS2に適用し、以下の論文に報告しました。
- Hikari Yoshitane*, Yoshimasa Asano, Aya Sagami, Seinosuke Sakai, Yutaka Suzuki, Hitoshi Okamura, Wataru Iwasaki, Haruka Ozaki, and Yoshitaka Fukada*. Functional D-box sequences reset the circadian clock and drive mRNA rhythms. Communications Biology (2019) DOI:10.1038/s42003-019-0522-3
MOCCSとは?
MOCCS(Motif Centrality Analysis of ChIP-Seq)は、様々なChIP-Seqのデータから転写因子が認識する配列を正確に抽出できる 新しいバイオインフォマティクス手法です。既存のモチーフ探索ツールが位置特異的重み行列(Position weight matrix)など要約された情報を抽出するのに対し、MOCCSは個々の配列がゲノム全体でどの程度認識されているかを評価します。
MOCCSについて、以下の論文にて報告しています。
- Yoshitane H*, Ozaki H*, Terajima H*, Du NH, Suzuki Y, Fujimori T, Kosaka N, Shimba S, Sugano S, Takagi T, Iwasaki W#, Fukada Y#. CLOCK-controlled polyphonic regulation of circadian rhythms through canonical and noncanonical E-boxes. Molecular and cellular biology. 2014 May 15;34(10):1776-87. (* Equal contributions, # Corresponding authors) 日本語プレスリリース
- Ozaki H, Iwasaki W. MOCCS: Clarifying DNA-binding motif ambiguity using ChIP-seq data. Computational biology and chemistry. 2016 Aug 1;63:62-72. Preprint
MOCCS2における改良点
MOCCSでは候補認識配列の転写因子結合への集中度合いを AUC という指標で評価しますが、候補認識配列の出現回数が小さい場合にAUCの値のばらつきが大きく偽陽性が生じうるという課題がありました。
MOCCS2では、そのようなk-merの出現回数に応じたAUCのばらつきの大きさを補正した ‘MOCCS2スコア’ を採用し、より偽陽性の少ない転写因子の認識配列を抽出することが可能になりました。MOCCS2では、全てのk-merについてAUCとMOCCS2スコアを計算していますが、MOCC2スコアを用いることを推奨しています。
アルゴリズムの概要
AUCによる認識配列の抽出
一般に、ChIP-Seqによって決定された転写因子結合サイト周辺での既知の認識配列の出現頻度を調べると、結合サイトを中心としたピーク様の分布を示します(図1)。MOCCSはこの性質を転写因子の認識配列の抽出に利用します。すなわち、転写因子結合サイト周辺での長さkの塩基配列(k-mer)の出現頻度がピーク様の分布となる配列を認識配列とし、全てのk-merの中から認識配列を列挙するというのがMOCCSのアイデアです。
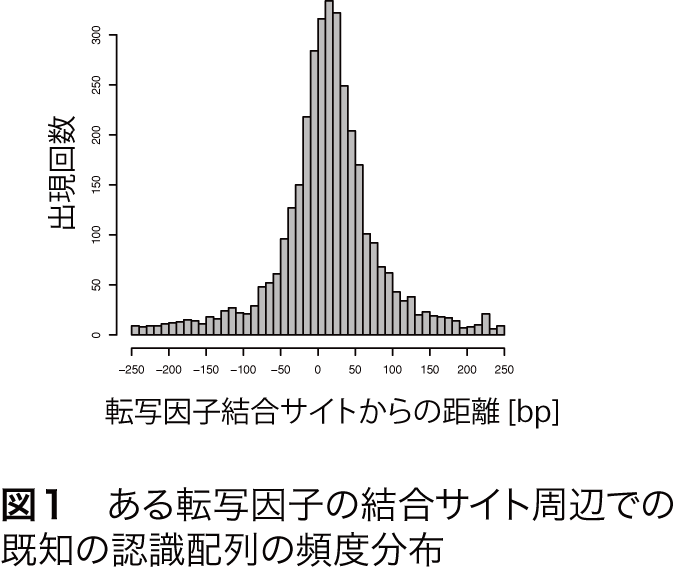
このピーク様の分布の鋭さを定量するために、まず、結合部位の中心からの距離を横軸に取り、累積の出現回数を縦軸に相対値としてプロットします(図2)。元の分布がピーク様の分布を示す場合、この曲線は上に凸の形になります(図2、赤線)。一方、ある配列が転写因子結合サイトの周辺にランダムに出現する場合は図2の青線のようになります。つまり、頻度分布のピークが鋭いほど、図2の赤線は青線から離れる形になります。この赤線と青線で囲まれた領域の面積をAUC(Area under curve)として定量すると、明確なピーク様分布を示す場合、AUCは大きくなることが分かります(図2)。
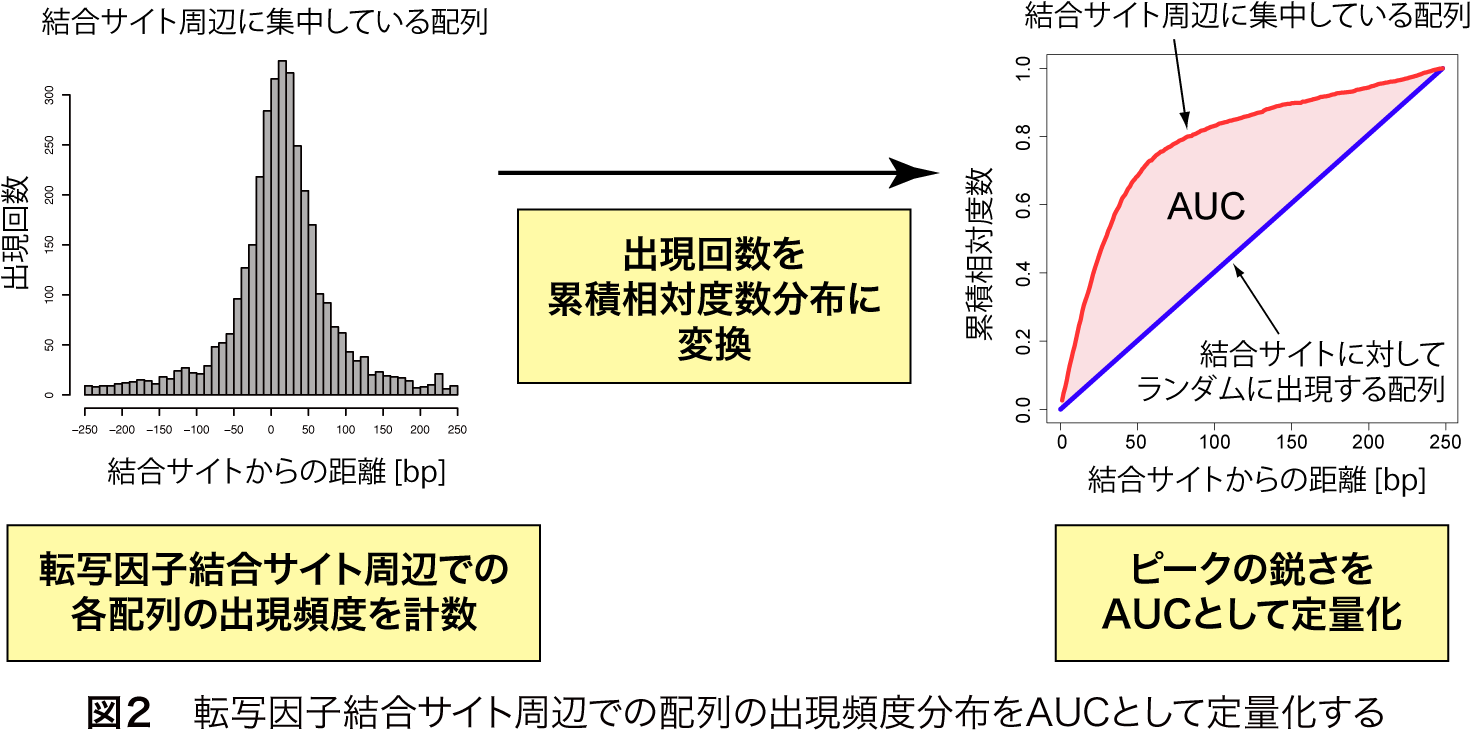
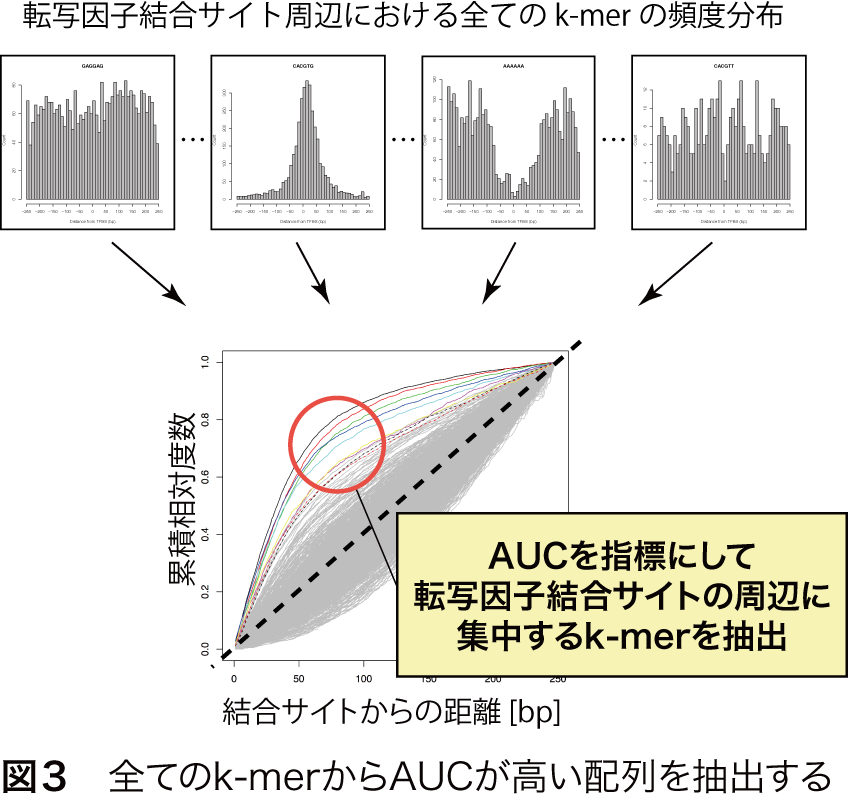
MOCCSは、全てのk-merについて、転写因子結合サイト周辺の頻度分布を調べ、AUCを計算し、AUCが閾値より高いk-merを列挙することで、転写因子の認識配列として抽出することができます(図3)。
MOCCS2スコアによるAUCのばらつきの補正
AUCはk-merの出現回数が小さいときに値が不安定になり、偽陽性が生じうるという課題がありました。
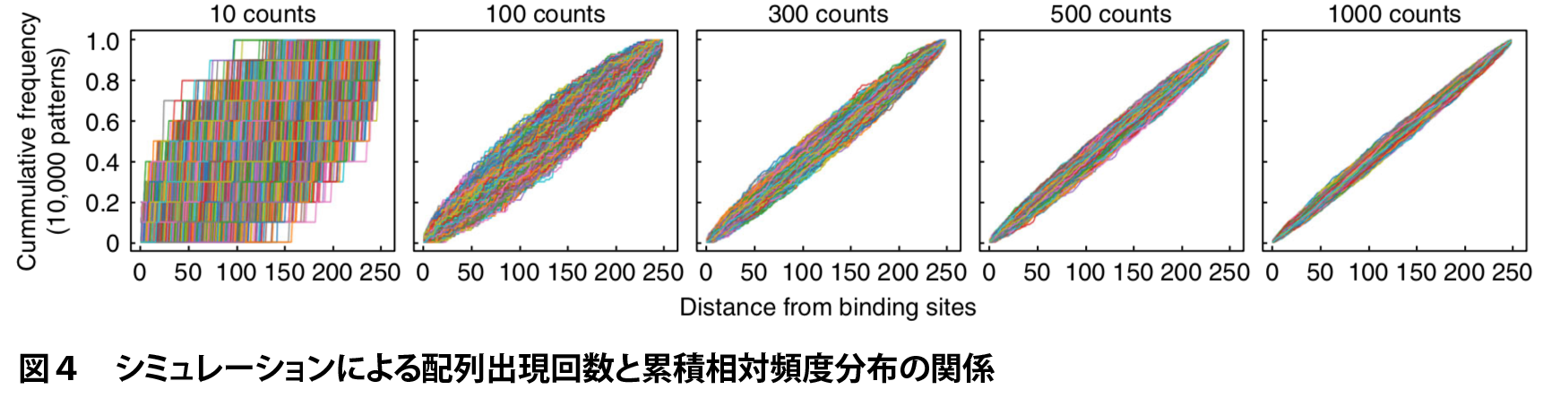
実際、転写因子結合サイト周辺に各k-merが一様分布から一定回数出現すると仮定してシミュレーションを行った場合、出現回数が大きいときは累積相対頻度分布が直線に乗るのに対し、出現回数が小さいときは直線からのばらつきが大きくなります(図4)。AUCの値のばらつきの度合いを標準偏差(SD)で定量すると、出現回数が小さいときは標準偏差が大きくなる、すなわち、AUCの値が大きなばらつきを示すことがわかります(図5)。
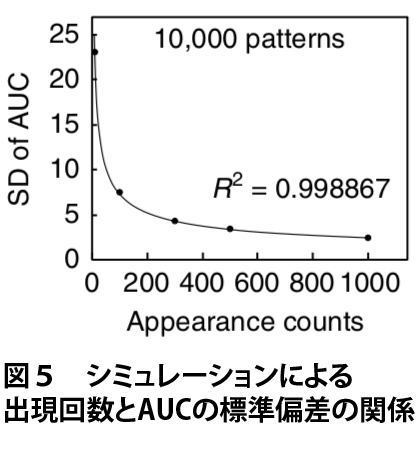
そこでMOCCS2では、そのようなk-merの出現回数に応じたAUCの標準偏差でAUCを補正した ‘MOCCS2スコア’ を計算することで、転写因子の認識配列を正しく抽出することを目指しました。
MOCCS2スコア は、あるk-merの出現回数におけるAUCの標準偏差に対する実際のAUCの相対値です。あるk-merの出現回数を とし、TFBS周辺のk-mer探索領域の長さを
とすると、MOCCS2スコアは以下のように定義されます:
AUCの標準偏差 [SD of AUC] の導出については、上記論文をご参照ください。
MOCCS2の使用例1: DBPおよびE4BP4 ChIP-seqデータの解析
ChIP-seq解析
まず、マウス肝臓由来の時計タンパク質であり転写活性化因子であるDBPと転写抑制因子であるE4BP4のChIP-seqデータから、両方の転写因子が結合する 1,490 の結合サイトを決定しました。
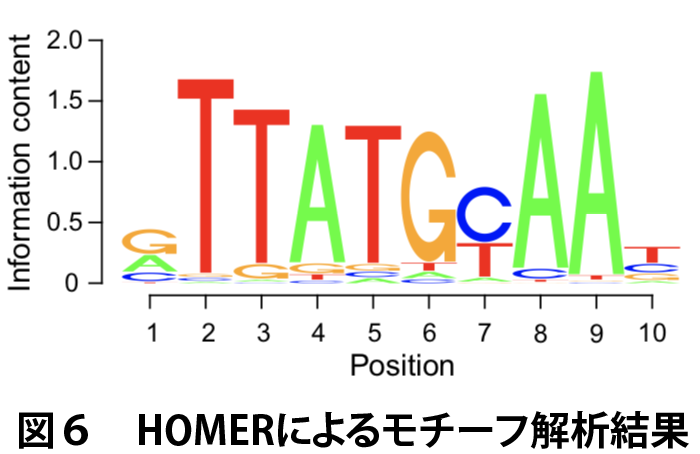
モチーフ探索ツールのHOMERを適用したところ、少なくとも長さ8 bpのモチーフがDBPとE4BP4に認識されることがわかりました(図6)。
MOCCS2解析
この結合サイト周辺の±250 bpの塩基配列を取り出し、MOCCS2解析に供しました。探索する認識配列の長さは5 bpから10 bpについて調べました。HOMERによるモチーフ解析の結果から、特に8 bpでのMOCCS2の結果に着目しました。
ここで、MOCCS2スコアが高い順に8 bpの配列を並べ替えた上で、MOCCS2スコアで上位にある配列と一塩基ずれで重なりのある配列(例えば、 TTATGCAA に対する TATGCAAN や NTTATGCA)は除外し、DBP/E4BP4認識配列を抽出しました(表1)。
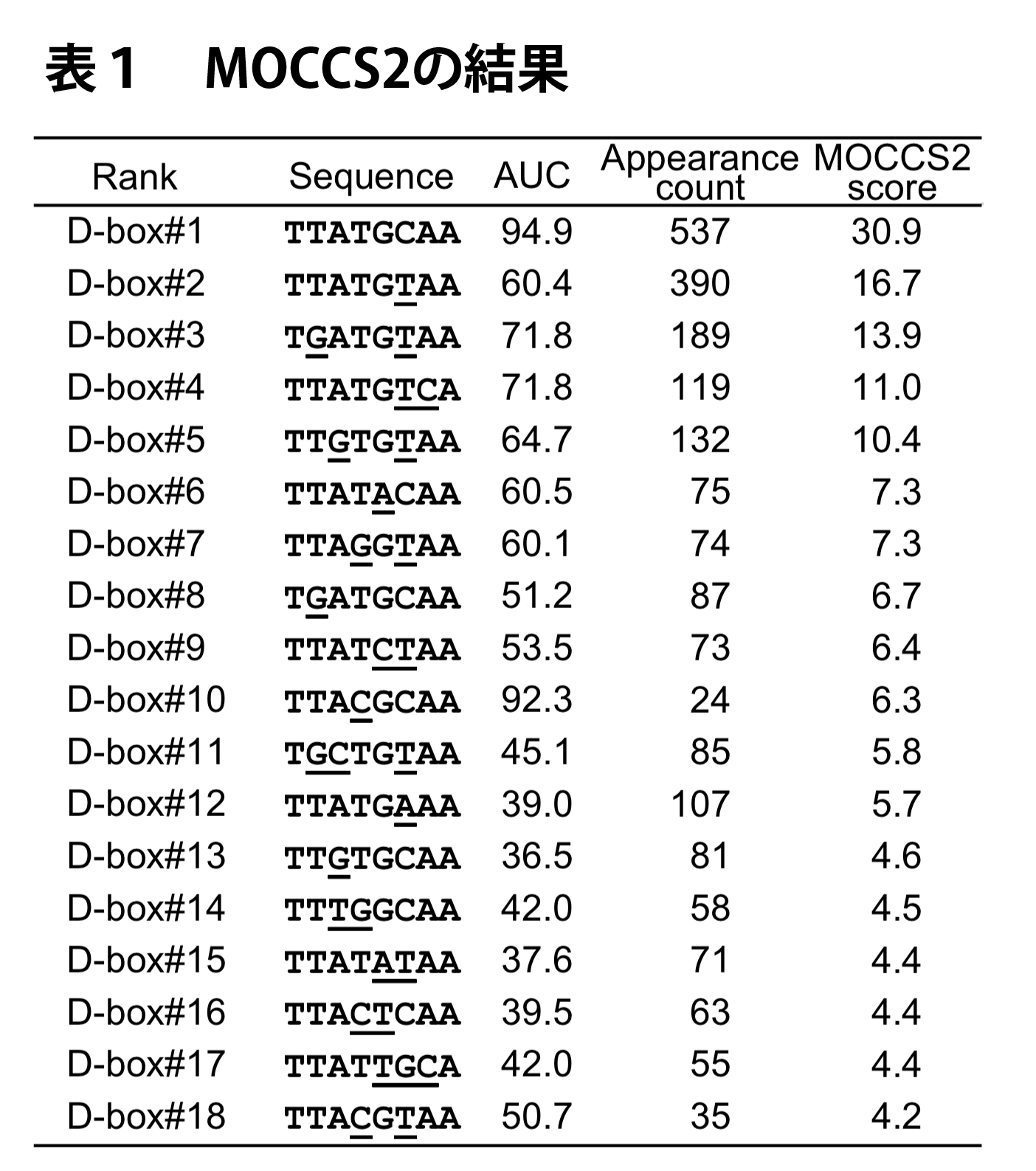
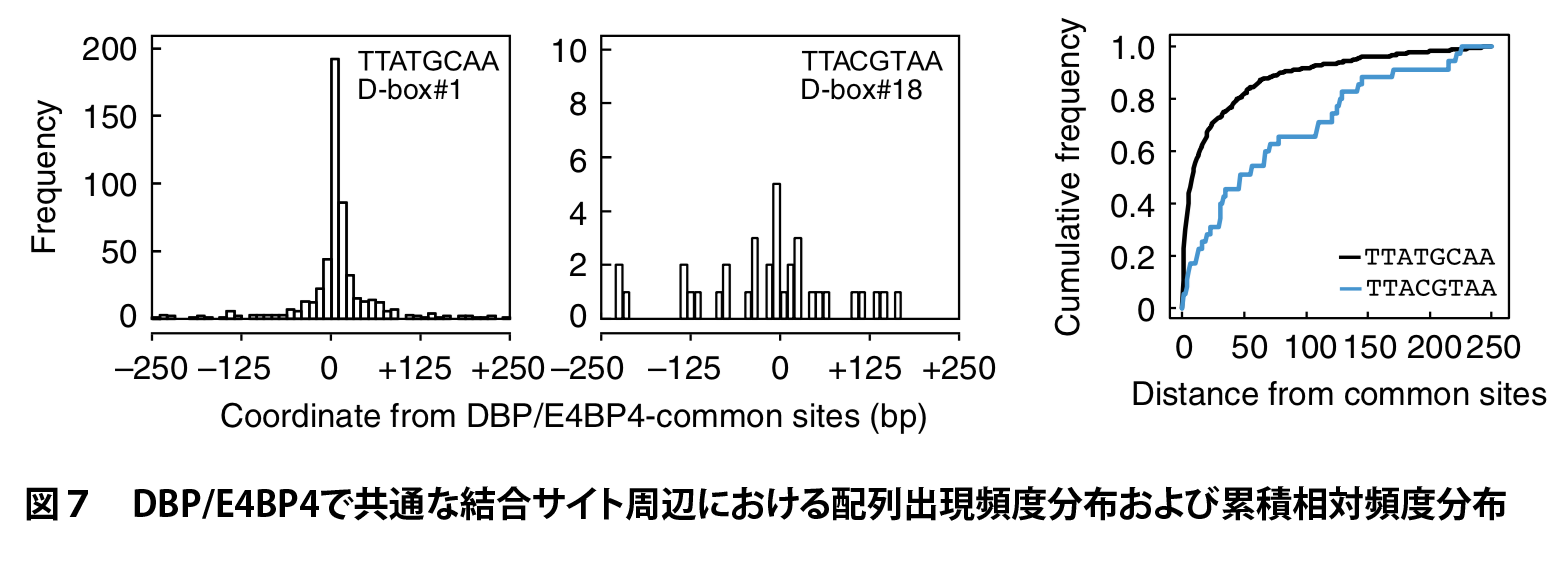
MOCCS2スコアが最も高かった配列は TTATGCAA でした。実際、DBP/E4BP4結合サイト周辺における TTATGCAA の分布をみるとピーク様の分布を示すことがわかります(図7)。この配列は Per1 遺伝子の転写開始点周辺において機能することが知られる認識配列であり、かつ、先行研究で知られているD-boxモチーフTTAYGTAAには含まれていなかったものでした。また、2番目の TTATGTAA は Per2 遺伝子のプロモーター領域におけるD-boxモチーフでした。さらに、18番目の TTACGTAA は D-boxモチーフ TTAYGTAA に含まれるものであり、出現回数は低いものの実際に結合サイト周辺に集中して分布していることがわかります(図7)。
実験による検証
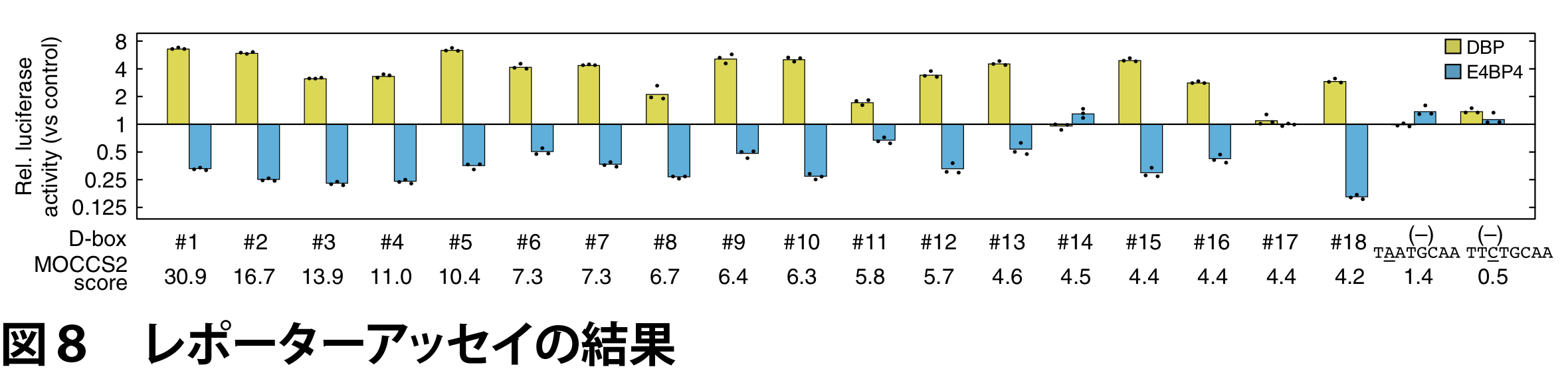
MOCCS2で抽出された認識配列が実際に機能を示すかを確かめるため、DBP依存的な転写活性化およびE4BP4依存的な転写抑制の誘導の有無をルシフェラーゼアッセイにより調べました。同時にネガティブコントロールとして、MOCCS2スコアでトップの TTATGCAA と一塩基ミスマッチであるもののMOCCS2スコアが低い配列(TAATGCAA、TTCTGCAA)でも同様の実験を行いました。
その結果、表1の多くの配列において機能が確認されました(図8)、一方、MOCC2スコアの低い配列では、転写活性化および転写抑制能が観察されませんでした(図8)。以上の結果より、MOCCS2によって実際に機能する認識配列を正確に予測できたことを表しており、MOCCS2の有用性を示すものです。
MOCCS2の使用例2: CLOCK ChIP-Seqデータの再解析
MOCCS解析
私たちは Yoshitane et al. (2014) において、CLOCK ChIP-Seqデータに対し、MOCCSを適用しました。まず、ピーク検出により、約8,000のCLOCK結合サイトを決定しました。この結合サイト周辺±250 bpの配列を解析に使用しました。
この研究では、CLOCKの典型的な認識モチーフであるCACGTGに類似した配列がどの程度ゲノム全体で使用されているかに興味があったため、探索する塩基配列の長さは6 bpとしました。また、CLOCKは遺伝子の転写開始部位の近傍に結合する傾向を示したため、マウスの全遺伝子のプロモータ配列(TSS上流501 bp)を用いて全ての6-merについてAUCを計算し、その分散の値を用いてAUCを正規化しました。
AUCの計算結果から、このようにすでにAUCで上位にある配列と重なりのある配列はを除外し、さらに正規化したAUCが5以上という基準を用いて、CLOCK認識配列を抽出しました(図9、表2)。
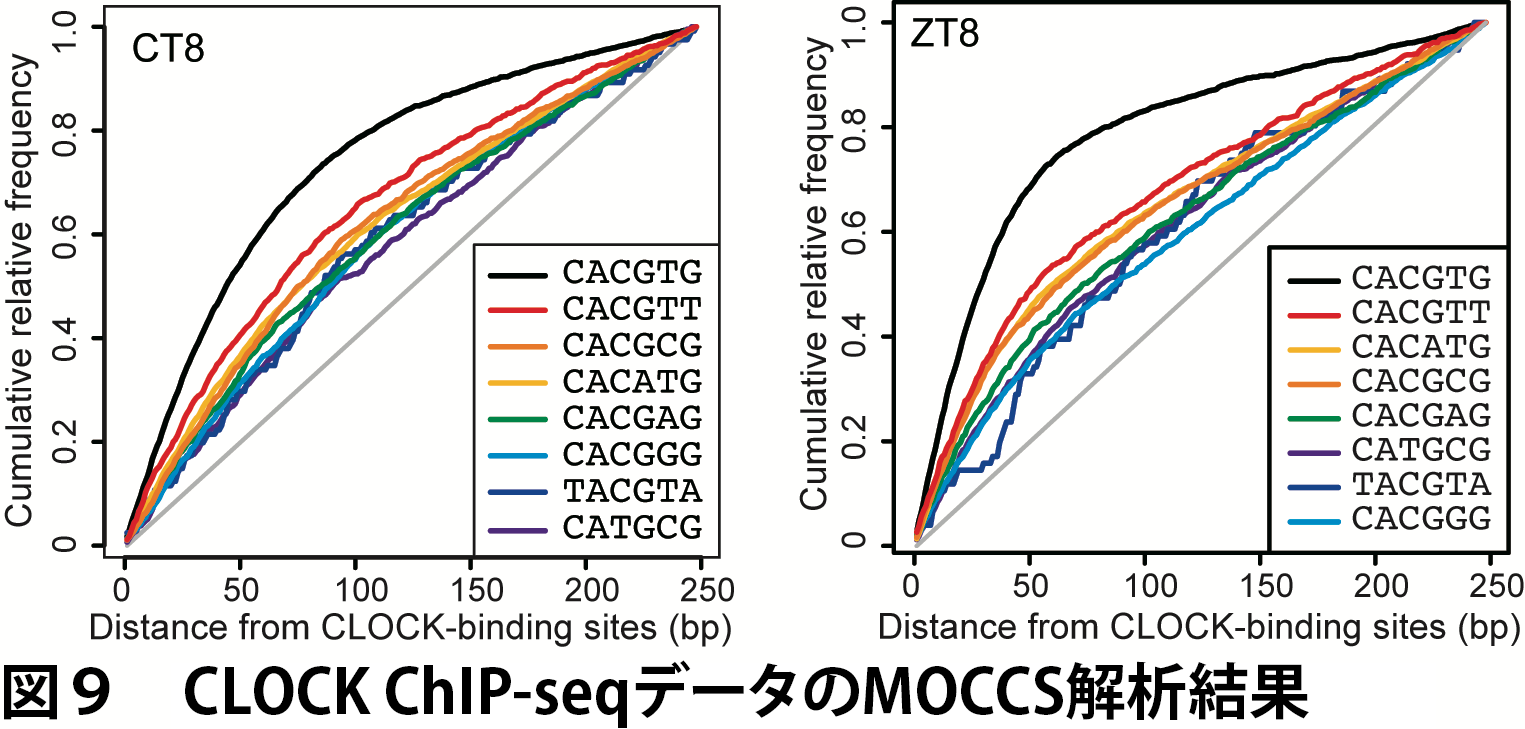
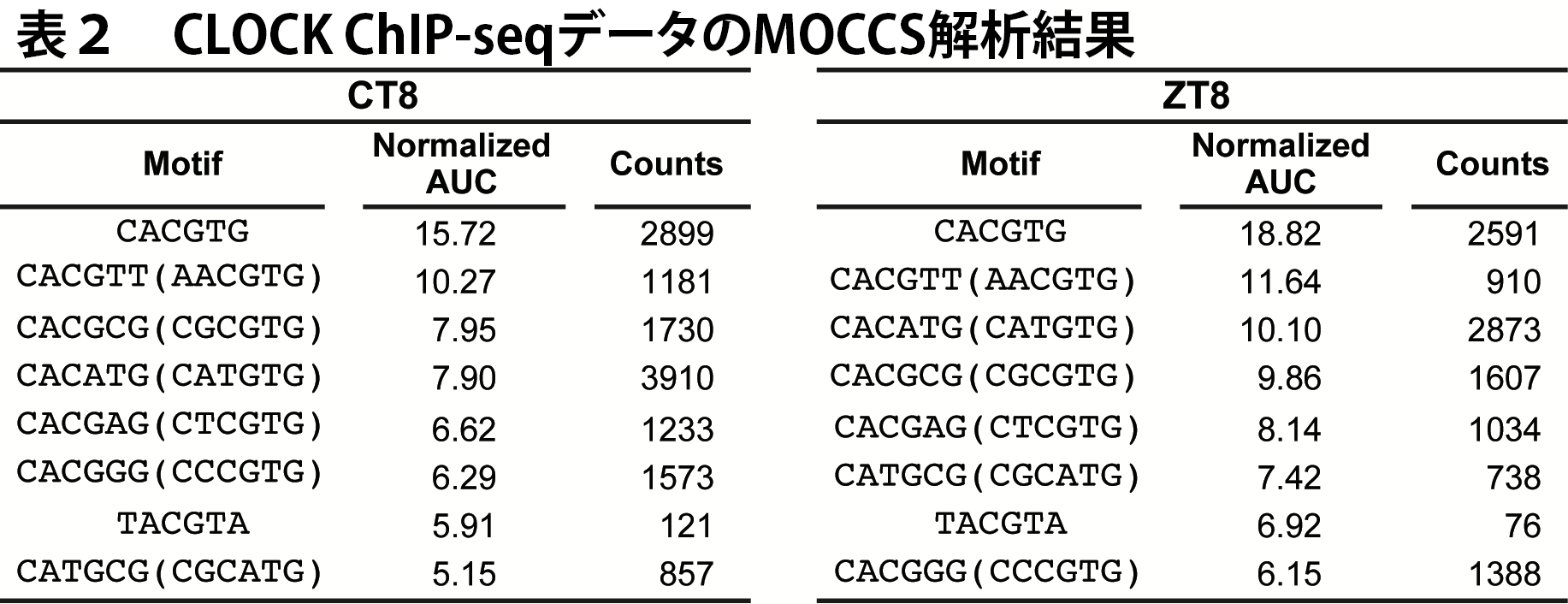
MOCCSの結果、AUCが最も高かったのはCACGTGでした(表2)。これは。CACGTGが最もよく使用される認識配列であることを示していて、MOCCSが先行研究の知見を再現できたと考えられます。また、CACG[ACGT]Gが全て抽出されたことから、ゲノム全体のレベルでは5番目(2番目)の位置は塩基についてあまり選択的でないことが分かりました。さらに、これまで知られていなかった、CACGTGと2塩基異なる配列が有意な認識配列として見つかりました。
実験による検証
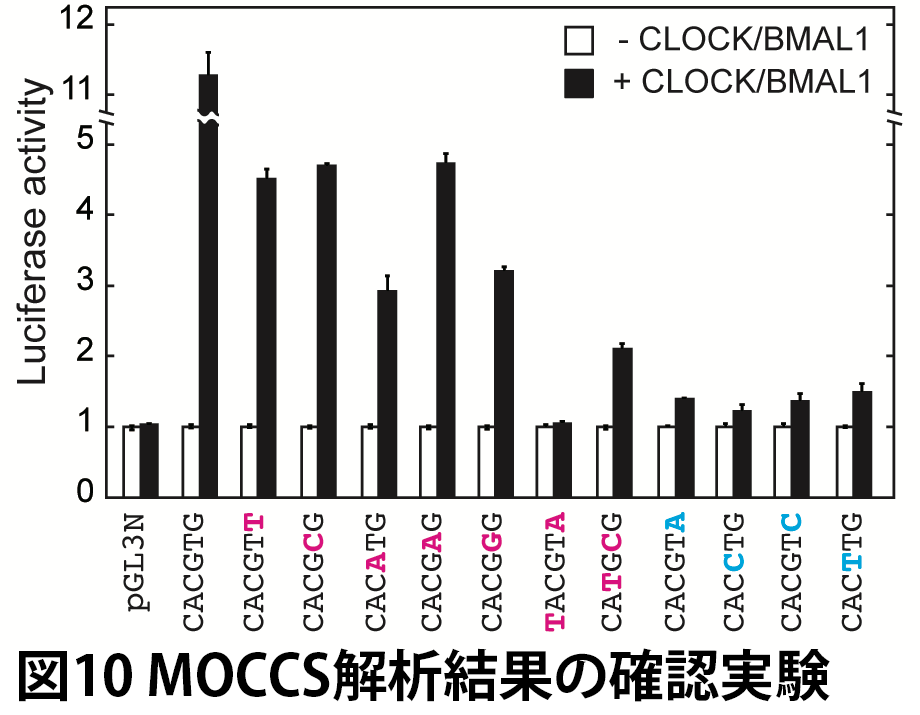
MOCCSで抽出された認識配列が実際に機能を示すかを確かめるため、CLOCK依存的な転写活性化の誘導の有無をルシフェラーゼアッセイにより調べました。その結果、表2のほぼ全ての配列において機能が確認されました(図10)。これは、MOCCSが実際に機能する認識配列を正確に予測できたことを表しており、MOCCSの有用性を示すものです。唯一機能を示さなかった TACGTA については、出現回数が少ないことによってAUCの値がノイズの影響を受けた可能性が考えられました。
MOCCS2による再解析
上記CLOCK ChIP-seqデータをMOCCS2によって再解析したところ、TACGTAについてのみAUCが高いにもかかわらずMOCC2スコアは低い値をとる結果となりました(図11)。これは TACGTA が出現回数が低いことによってAUCの値が不安定なことによる偽陽性であることを示すものであり、レポーターアッセイ実験の結果とも整合的です。
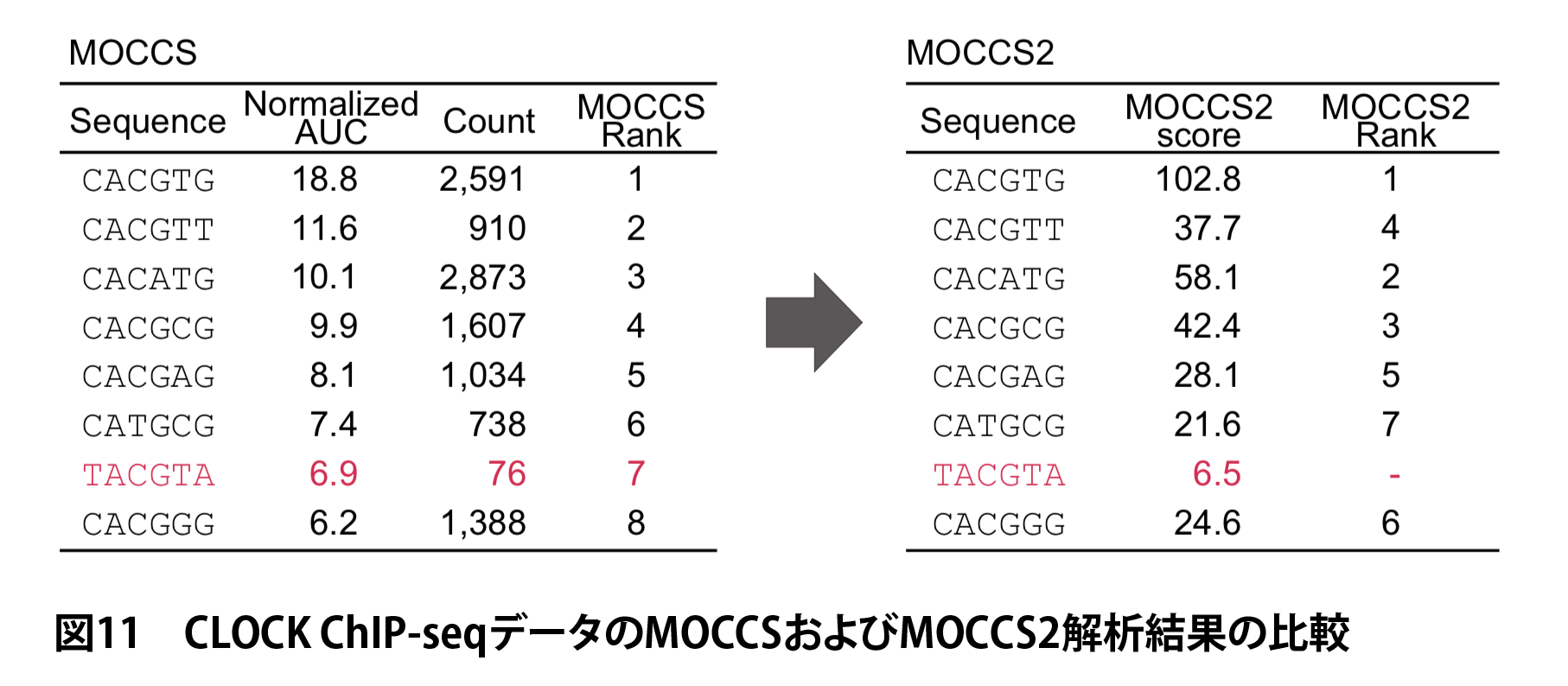
この結果から、MOCC2スコアは実際にAUCのばらつきによる偽陽性を除き、より正確に転写因子の認識配列を列挙できることが示されました。
連絡先
MOCCS2は様々なChIP-Seqデータから有意なDNA結合タンパク質の認識配列を列挙することができる手法です。興味のある方は 尾崎 harukao.cb[at]gmail.com までメールにてご連絡ください。